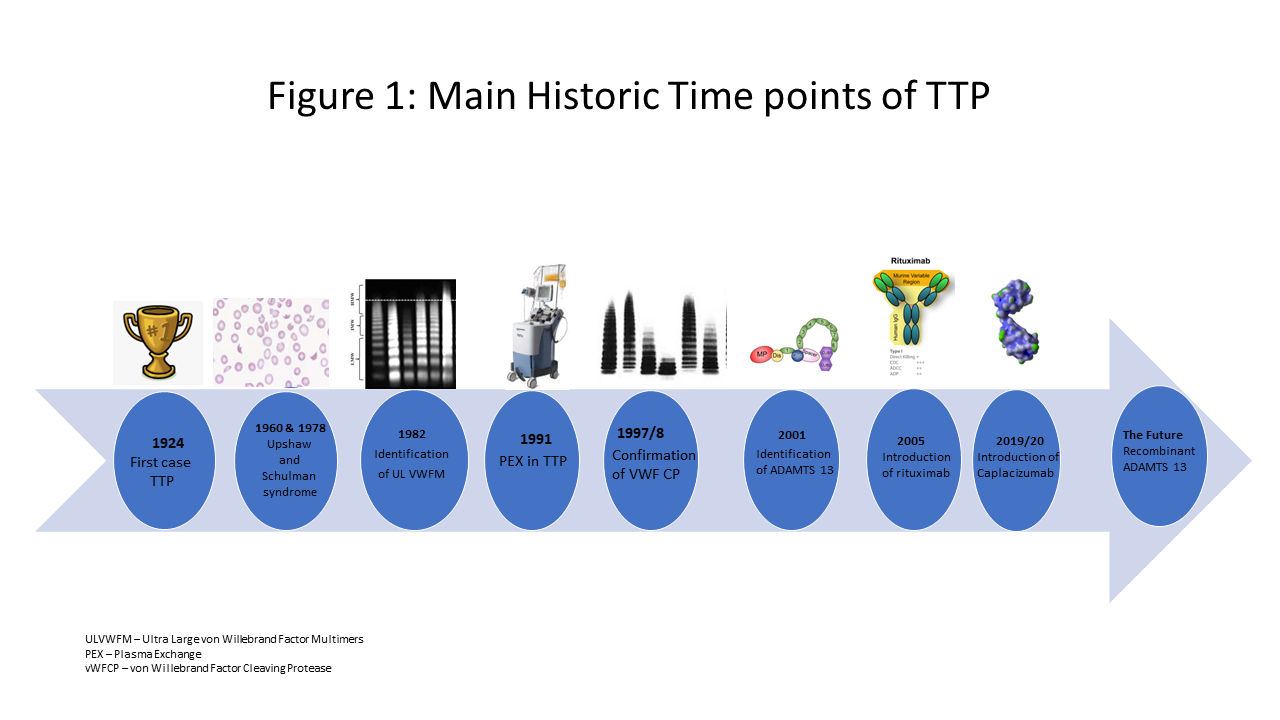
Thrombotic thrombocytopenic purpura (TTP) was first described in 1924 by Moschcowitz,1 who presented a fatal case of TTP associated with multi-organ failure over two weeks. Professors Upshaw (1978)2 and Schulman (1960)3 separately described cases we now recognise as congenital TTP, suggesting a deficiency in plasma and response to plasma infusion.
In 1982, Professor Moake4 identified the presence of ultra large von Willebrand Factor (VWF) multimers, specific to TTP. In 1997–1998, Professors Furlan5 and Tsai6 confirmed the absence of a protein in the plasma, named VWF-cleaving protease in acute and chronic relapsing TTP cases. In 2001, Professor Gallia Levy confirmed this protein was a metalloprotease named ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) (Figure 1).7
Diagnosis
Central to the diagnosis of TTP is the presence of thrombocytopenia, a haemolytic anaemia with specific blood film changes, specifically schistocytes or fragmented red cells. The condition is an acute, life-threatening medical emergency, and symptoms relate to microvascular thrombi with associated end organ damage. Timing of diagnosis and treatment is critical and the heterogenous presentation may make this challenging. However, new thrombocytopenia requires a blood film analysis.
TTP occurs in women in approximately 70% of cases and the median age of presentation is 40-years-old, but it can present in young children or the elderly. The incidence is estimated as one to two per million per year with a prevalence of ten per million.8 Confirmation of the diagnosis is the demonstration of a severe deficiency of the enzyme, ADAMTS13 (less than 10%).
ADAMTS13 is important in the cleavage in the A2 region of VWF as it is secreted from endothelial cells. This physiological action generates more haemostatically active VWF multimers. VWF multimers bind with platelets and this action is important in primary haemostasis. However, in TTP, the lack of ADAMTS13 generates an increase in ultra large VWF multimers that promote increased platelet binding and aggregation. These break off as microthrombi and result in organ damage (Figure 2).
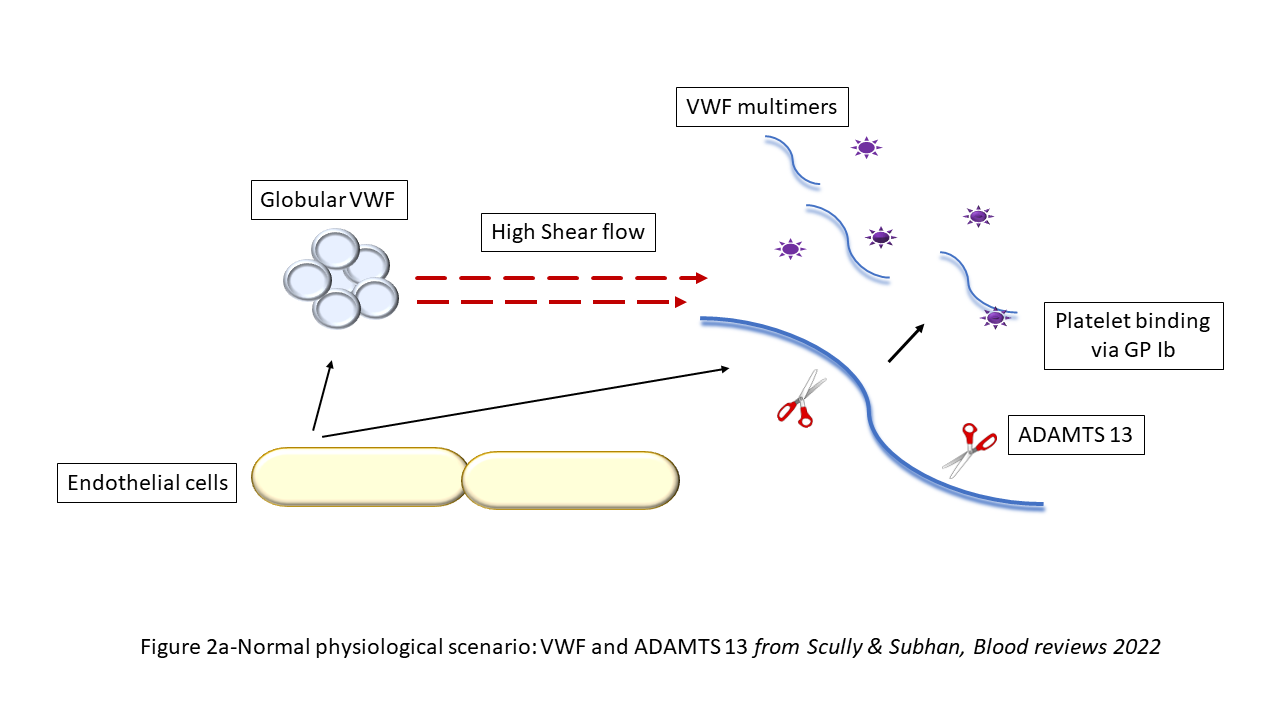
Figure 2a. Normal physiological scenario: VWF and ADAMTS 13
Reprinted from Blood Reviews, 55, M Subhan & M Scully, Advances in the management of TTP, 100945, © Elsevier, 2022, with permission from Elsevier.
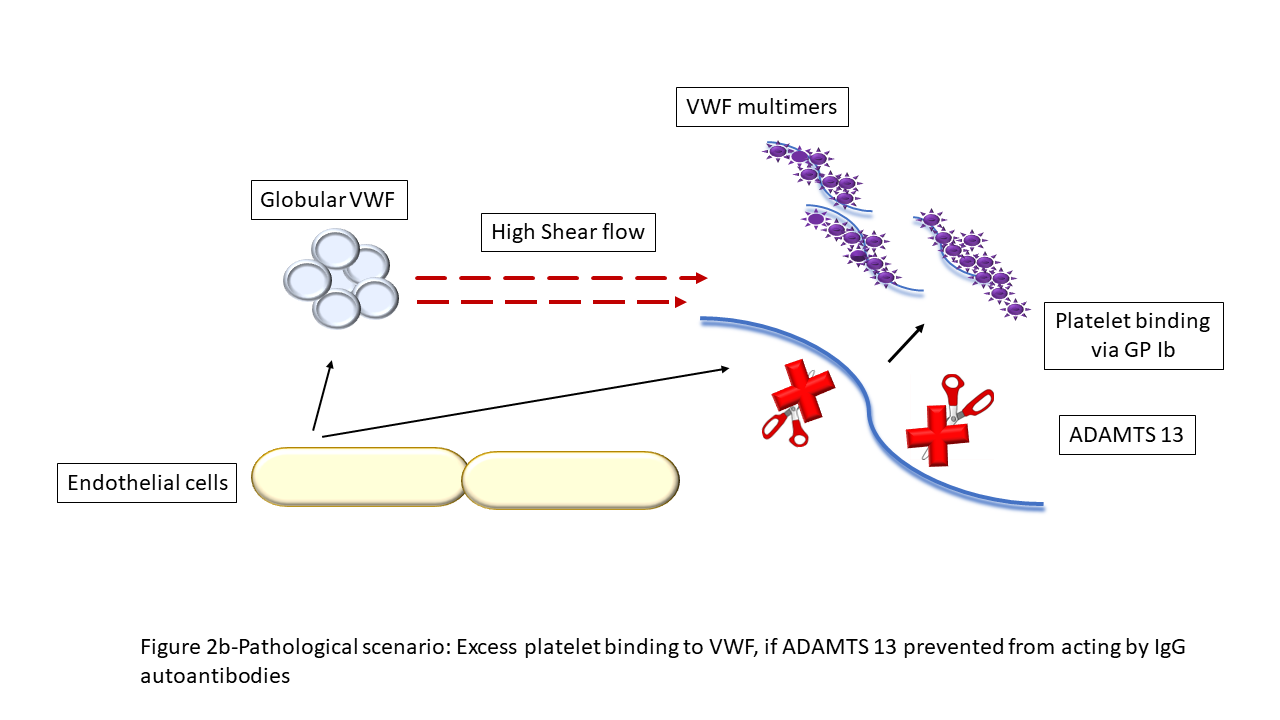
Figure 2b. Pathological scenario: Excess platelet binding to VWF, if ADAMTS 13 prevented from acting by IgG autoantibodies.10VWF: von Willebrand factor.
Reprinted from Blood Reviews, 55, M Subhan & M Scully, Advances in the management of TTP, 100945, © Elsevier, 2022, with permission from Elsevier.
TTP is subclassified into immune mediated TTP (iTTP, 90% of cases), associated with antibodies to ADAMTS13, and congenital TTP (cTTP, approximately 5% of cases), caused by mutations within the ADAMTS13 gene, inherited in an autosomal recessive manner. In the remaining cases, there is a precipitating cause related to iTTP, such as pregnancy, HIV or drugs.
The past
Untreated, the mortality of TTP is over 90%. The mainstay of treatment is plasma exchange (PEX) which allows a large volume delivery of ADAMTS13. Indeed, its benefit was shown by the Canadian Apheresis Group study comparing PEX to plasma infusion (PI) in 1991.9 This demonstrated the survival benefit of PEX to 80%. However, there remained significant morbidity and relapses.
Pathology has been central to the progress of TTP management, particularly in the UK, via the work of clinical haematologists, laboratory biomedical scientists, specialist haemostasis laboratories and multicentre national research.
Steroids have been used since the 1980s to positive benefit in patient survival when used in conjunction with PEX, but they do not reduce relapse rates. A number of other immunosuppressive therapies were used, often associated with toxicity. The introduction of rituximab, the anti-CD20 monoclonal antibody, in the mid-2000s was the next therapeutic turning point in TTP care. Following a regimen comparable to that used in lymphoma treatment, rituximab use in acute refractory and relapsing TTP demonstrated clearance of anti-ADAMTS13 IgG antibodies and normalisation of ADAMTS13 activity levels. Earlier use of rituximab in the acute treatment pathway (within three days of admission) reduced inpatient stay to a median of 14 days (21 days if admitted to the intensive care unit). Furthermore, the time to subsequent relapse was reduced from 50% within 18 months to 15%.
The utility of rituximab in normalising ADAMTS13 activity levels has resulted in it being used to prevent TTP relapses. Monitoring of ADAMTS13 activity levels, and administration of rituximab when ADAMTS13 activity is reduced to 15–20% from normal, prevents acute TTP relapses.10
The present
Since 2019, following publication of the phase III international multicentre trial,11 the nanobody caplacizumab has been used as standard of care in the acute TTP pathway in the UK. Caplacizumab is an anti-VWF antibody and binds to the A1 region of VWF, specifically on the platelet binding receptors. When given to patients with acute TTP, this results in a quicker time to platelet normalisation, which is sustained, preventing exacerbations and refractory disease. Caplacizumab does not affect the underlying ADAMTS13 axis, but is an important adjunct therapy, thereby protecting patients and maintaining clinical remission pending response to immunosuppressive therapy. Addition of caplacizumab has reduced further length of inpatient admission and, if used promptly, there is a reduction in mortality and morbidity. Although caplacizumab action on VWF results in a severe von Willebrand disease state, clinically significant bleeding is rare given the prothrombotic nature of TTP.
The current treatment pathway in acute TTP is prompt transfer to the regional TTP centre and initiation of PEX, preferably within 4 hours whenever TTP is suspected clinically.12 Confirmation of TTP diagnosis via ADAMTS13 activity assay should be made within 24 hours, but this should not delay treatment.
PEX is undertaken using Octaplas (Octapharma, Austria), a double viral inactivated fresh frozen plasma (FFP) with a prion reduction step. Its benefit over standard FFP relates to pathogen reduction and very low rates of reactions, a significant factor when the median volume of plasma in an acute TTP episode is 40 litres. Other key components of acute therapy are immunosuppression with steroids and rituximab, and early use of caplacizumab (Figure 3).
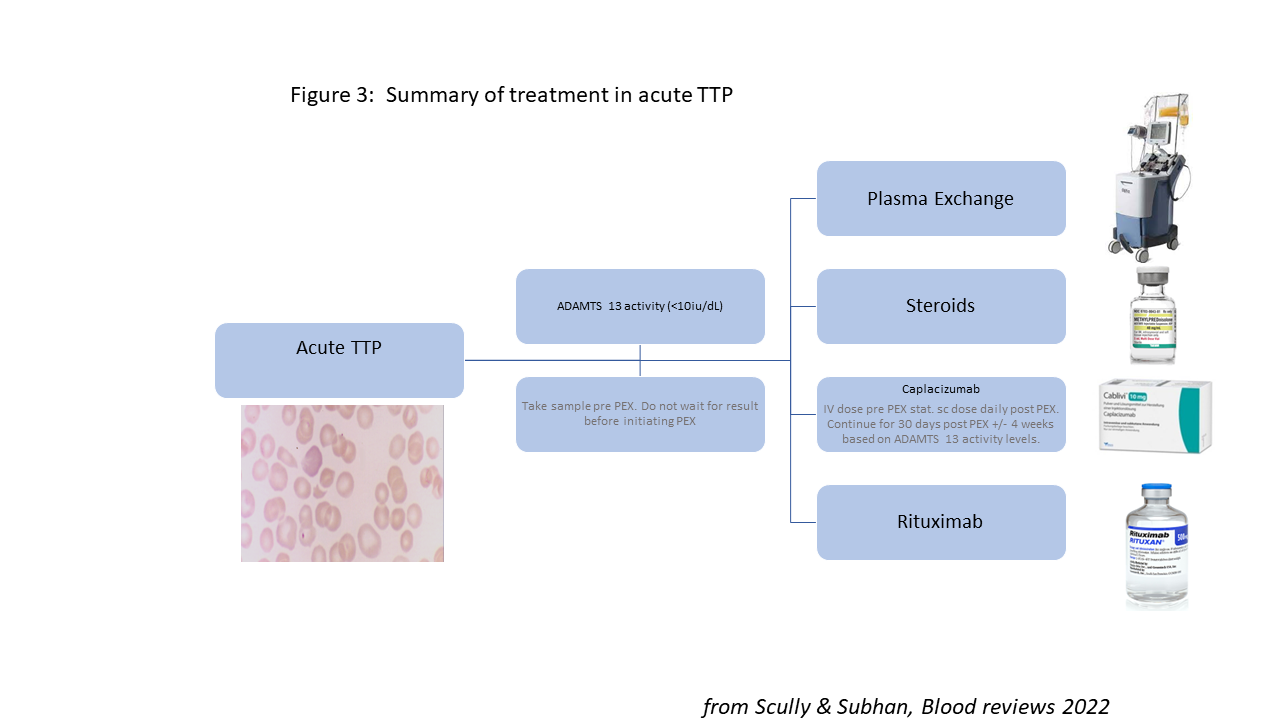
Reprinted from Blood Reviews, 55, M Subhan & M Scully, Advances in the management of TTP, 100945, © Elsevier, 2022, with permission from Elsevier.
The incidence of cTTP is less than one per million per year. It is now clear that most cases occur in adults, mainly women in pregnancy, and without treatment it is associated with fetal loss. Management in subsequent pregnancies is with ADAMTS13 replacement, currently with plasma infusion throughout pregnancy and the post-partum period. Indeed, the emphasis is now to treat all cTTP patients from diagnosis to prevent end organ damage (the risk of stroke in those over 40-years-old and not on treatment is 50%13).
Even with therapy maintaining the platelet count in the normal range, patients with cTTP may suffer symptoms; these include prolonged headaches/migraines, abdominal pain or severe lethargy. Increasing the frequency of ADAMTS13 replacement can reduce or ameliorate these symptoms.
The role of pathology
TTP requires a multidisciplinary team approach to care. Pathology has been central to the progress of TTP management, particularly in the UK, via the work of clinical haematologists, laboratory biomedical scientists, specialist haemostasis laboratories and multicentre national research.
The future
TTP is designated a highly specialised service and commissioned at a regional level within England to provide dedicated care in appointed units. This will provide equity of care, long-term follow-up and standardised treatment approaches, aiming to reduce mortality to less than 10% and relapse rates to under 15%. The future availability of the missing ADAMTS13 protein in a recombinant form will allow personalised treatment approaches for cTTP cases and ultimately avoid the need for PEX in iTTP.
Author(s)

Marie Scully