Since the initial drug received Food and Drug Administration (FDA) approval in the USA in 2017, CAR-T therapy has emerged as one of the biggest breakthroughs in cancer therapy in decades. Often described as a ‘living drug’, the treatment has been funded by NHS England since 2018 and several products are now used routinely or in trials to treat a range of haematological and non-haematological malignancies. Here, we give a brief overview of CAR-T therapy and its application in clinical practice.
CAR-T therapy explained
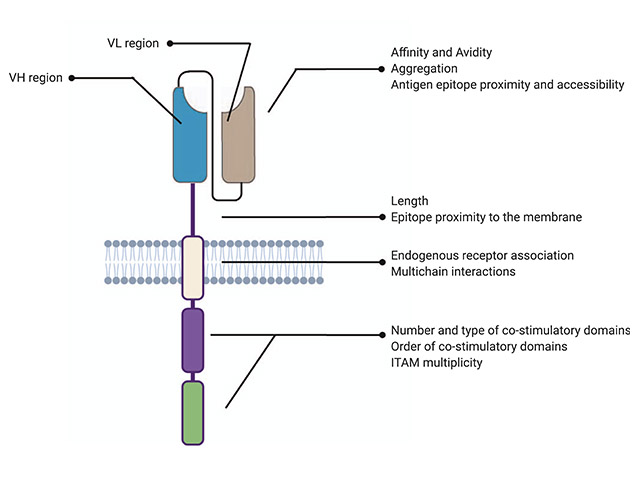
The CAR-T cell drug is created by inserting a manmade gene, the chimeric antigen receptor (CAR), into the genomic DNA of lymphocytes using a lentiviral vector. The CAR consists of the following four separate regions, which together give the engineered cell the ability to exhibit both antigen binding and T-cell activating functions (Figure 1):1
- extracellular antigen recognising domain – usually derived from monoclonal antibody variable regions selected to target a certain surface marker, such as CD19 on B-cells
- hinge region – provides the antigen recognising domain with better physical flexibility to bind antigens
- transmembrane domain – links extracellular and intracellular parts
- intracellular signalling domain – varies between products but consists of part of the T-cell receptor complex, usually CD3 zeta, as well as one or more co-stimulatory molecules, such as CD28 or 4-1BB.1
Evidence and FDA approval
The FDA approved the first CAR-T therapy, tisagenlecleucel (Kymriah), in 2017 after trials in patients with relapsed and refractory B-cell acute lymphoblastic leukaemia (B-ALL) showed remarkable results.
A phase I/IIA study in 25 children and five adults with B-ALL demonstrated an exceptional complete response (CR) rate of 90%.2 This included high CR rates in patients with previous allogeneic stem cell transplants and other CD19-targeted therapies, such as blinatumomab. These results were revolutionary in this setting, which is usually characterised by limited treatment options and poor outcomes. Unfortunately, early relapses remained an issue.
Following this, the ELIANA study, an international phase II study of 75 patients with multiply relapsed B-ALL, achieved excellent CR rates of 81% and more durable responses with six months event-free and overall survival rates of 73% and 90%, respectively. The median duration of remission was not reached at the end of the study period of 20 months and CAR-T cells were detectable for the duration of data collection.3 The success of this study led to FDA approval in B-ALL and further trial roll out across other haematological malignancies.
The pivotal JULIET and ZUMA studies, for tisagenlecleucel and axicabtagene ciloleucel (Yescarta), respectively, led to approval in diffuse large B-cell lymphoma (DLBCL) shortly after. Initial trial outcomes are summarised in Table 1.4
The first two products, tisagenlecleucel, with licence in B-ALL, and axicabtagene ciloleucel with licence in DLBCL, have been routinely available in the UK for several years,5 with others including brexucabtagene autoleucel (Tecartus) in mantle cell lymphoma and lisocabtagene maraleucel (Breyanzi) in DLBCL having recently been approved.
Challenges of CAR-T therapy
Despite encouraging results from clinical trials and significant progress in CAR-T research, several important challenges remain.
| Table 1: Reported clinical efficacies of CAR-T therapies (includes data on ELIANA, JULIET, ZUMA-1, ZUMA-2 and TRANSCEND). | ||||||
| CAR-T therapy | Disease | Number of patients who received CAR-T | CR (%) | OS (%) | DFS/R (%) | Reference |
| Tisagenlecleucel | ALL | 30 | 90 | 78 | 67 (6 months) | 2 |
| Tisagenlecleucel | ALL | 75 | 81 | 90 (6 months) | 73 (6 months) | 3 |
| Tisagenlecleucel | Large B-cell lymphomas | 93 | 40 | 49 (12 months) | 65 (12 months) | 6 |
| Axicabtagene ciloleucel | Large B-cell lymphomas | 101 | 54 | 52 (18 months) | 44 (12 months) | 7 |
| Brexucabtagene autoleucel | Mantle cell lymphoma | 68 | 67 | 83 (12 months) | 61 (12 months) | 8 |
| Lisocabtagene maraleucel | Large B-cell lymphomas | 269 | 53 | 75 (6 months) | 51 (6 months) | 9 |
| ALL: Acute lymphoblastic leukaemia; CR: Complete response; DFS/R: Disease-free survival/remission; OS: Overall survival. | ||||||
Loss of target
As CAR-T therapy is targeted to a specific surface antigen, cancer cells that reduce or even lose expression of the target antigen (e.g. CD19) will become invisible to the treatment. This is termed antigen escape and has been demonstrated in 30–70% of patients with relapsed disease.10 Strategies to avoid antigen escape include development of dual-target CAR-T cells, which are undergoing clinical trials.
On-target-off-tumour toxicity
Choice of the target is vital as the CAR-T cell will attack any cell expressing the target antigen. This can cause significant issues, such as cardiac toxicity or B-cell aplasia when CD19 targets are used.10
Some of these difficulties may be overcome through use of allogeneic, off-the-shelf CAR-T products, which would be readily available at the required point in the patient’s treatment.
Manufacturing process
Using autologous cells as a bespoke product has resulted in large-scale logistical issues to collect, cryopreserve and transport cells to manufacturing centres around the world before returning them to the treating centre. The process takes a minimum of three to four weeks which poses significant challenges for many patients with rapidly progressive disease and often necessitates use of ‘holding’ chemotherapy to keep the disease under control. In a small proportion of cases, insufficient cells are collected or the manufacturing process may fail due to difficulties in T-cell selection and expansion.11 In a recent real-life study, only 57 of 122 patients had received CAR-T cells as planned with a further 45 awaiting leukapheresis or cell infusion. 18 did not proceed due to disease progression (n=16) or manufacturing failure (n=2), with two patients being monitoring clinically following radiotherapy and bridging chemotherapy.12
Some of these difficulties may be overcome through use of allogeneic, off-the-shelf CAR-T products, which would be readily available at the required point in the patient’s treatment.
While techniques are improving, there is still a large amount of progress needed to generate a successful product.
Therapy-related adverse effects
While CAR-T therapy has led to excellent results in some patients, treatment remains associated with significant and complex adverse events. Careful patient selection is essential to ensure fitness for therapy. The characteristics of each CAR-T cell drug differs even with the same target and therefore the toxicity also varies significantly. Non-relapse mortality for CAR-T therapy ranged between 0 and 8% in initial trials,13 and was reported as 6% in a recent real-life study.14
The best characterised and most common side effect is the cytokine release syndrome (CRS), which usually occurs in the first two to ten days after infusion. CRS is a systemic inflammatory response syndrome caused by the supra-physiological activation of pro-inflammatory cytokines, such as IL-2, IL-6, IFN gamma and TNF alpha, in response to T-cell activation.15 Patients may present at the milder end of the spectrum with fevers, myalgia or tachycardia but can progress rapidly to exhibit signs of hypoxia, capillary leak and hypotension which can lead to multiorgan failure and death.
As cytokine production is an essential part of T-cell activation, a degree of CRS is likely unavoidable and has been reported across all trials. CRS frequently requires intensive care support and is now routinely treated with the anti-IL6 antibody tocilizumab.16
A less well-understood side effect of CAR-T therapy is the immune effector cell neurotoxicity syndrome (ICANS). ICANS has been linked to cerebral oedema, microhaemorrhages, parenchymal necrosis and high levels of proinflammatory cytokines in the brain tissue but exact mechanisms are currently unclear. Patients can present with a wide range of neurological symptoms but most commonly exhibit headaches, tremors, signs of confusion, speech impairment (e.g. expressive dysphasia) and reduced levels of consciousness.17 Importantly, ICANS can progress rapidly from mild headache to coma, making early recognition essential.
To this end, neurocognitive assessments using immune effector cell encephalopathy scores are performed several times a day on all patients. CAR-T therapy is also associated with often profound cytopaenias caused by conditioning chemotherapy as well as the CAR-T treatment itself. While blood counts recover by one month in the majority, it should be noted that infection risk is high, necessitating broad spectrum antibiotic therapy in almost all patients.18
Finally, the true long-term effects of CAR-T therapy are not yet known as most trials are relatively recent. However, data is maturing and reports of ten-year remissions with limited late adverse events are emerging.19
Future directions
There is no doubt that CAR-T therapy has revolutionised the management of haematological malignancy for an increasing number of patients. We are seeing complete responses and even durable remissions in a landscape that was previously characterised by abysmal outcomes. Additionally, the improved management of toxicities with agents such as tocilizumab is leading to better clinical applicability. Despite this, significant challenges remain and work to improve products and practices is ongoing.
In the future, we are likely to see allogeneic, off-the-shelf CAR-T products which may overcome production failures and manufacturing time delays, but may also cause new challenges, such as graft versus host disease and cell product rejection.
Despite significant developments in this novel therapy, much remains unknown, and we will need to further our understanding of the intricacies of CAR design, manufacturing processes and product delivery.
Trials are additionally exploring the use of CAR-T therapy in other haematological malignancies, as well as in solid tumours such as neuroblastomas.
From a logistical perspective, regulators and clinicians are facing many challenges caused by the sometimes-bewildering pace of change in the field of CAR-T therapy. Providers will have to adapt to increasing patient numbers and will have to streamline processes to accommodate demand for CAR-T therapy. Additionally, the cost of treatment remains an issue and may lead to difficulties with access and exacerbate healthcare inequalities.
Despite significant developments in this novel therapy, much remains unknown, and we will need to further our understanding of the intricacies of CAR design, manufacturing processes and product delivery.
It is certainly an exciting time to work in cancer immunotherapy. It is now up to us to find the best way of utilising our increasingly complex living drugs by selecting the right drug at the right time and for the right patient to ultimately optimise care for even the most difficult to treat patients.
Author(s)

Dr Julia Wolf